Open-source AI-powered
drug design platform
From protein target to druggable pocket to predicted binding affinity — in minutes, not months. Built on AlphaFold 3, Boltz-2, P2Rank, and the latest open-source computational biology tools.
Built with leading open-source tools
in AlphaFold DB
prediction engines
databases integrated
MIT license
The Challenge
Developing a new drug takes 10–15 years and costs $2.6 billion
Only 12% of drugs that enter clinical trials are eventually approved.
OpenDDE accelerates the earliest stages — target validation, pocket discovery, and hit identification — where computational tools can save years of experimental work.
Drug Discovery 101
How drug discovery works
Understanding the journey from disease biology to approved medicine.
Identify the target
Every disease is caused by specific proteins malfunctioning in the body. The first step is identifying which protein to target — like finding which broken part of an engine to fix.
Find the binding pocket
Proteins have specific pockets on their surface where small molecules can bind, like a key fitting into a lock. Finding these pockets is critical — you need to know WHERE on the protein a drug can attach.
Discover candidate molecules
Researchers search databases of known compounds to find molecules that might fit the pocket. They look at what’s already been tested, analyze structure-activity relationships, and identify promising starting points.
Predict how drugs bind
Before testing in a lab, computational tools predict how a drug molecule will sit inside the pocket — its orientation, molecular contacts, and the strength of the interaction. OpenDDE pairs AlphaFold 3 for binding poses with Boltz-2 for predicted affinity (pIC50, IC50, binder probability), so you can both visualize and rank.
Optimize and test
The best candidates are refined — modified to bind more tightly, be more selective, and have fewer side effects. Then they enter lab testing and eventually clinical trials.
The Impact
Why computational drug design?
AI predicts binding in seconds vs weeks of wet-lab experiments
Virtual screening replaces months of manual compound testing
Discover pockets on previously "undruggable" proteins
“[IsoDDE] more than doubles the accuracy of the best existing method, AlphaFold 3, in predicting binding poses for drug-like molecules.”
OpenDDE brings these capabilities to every researcher, for free, using open-source tools. No cloud GPU required. No vendor lock-in. Just science.
Platform
See OpenDDE in action
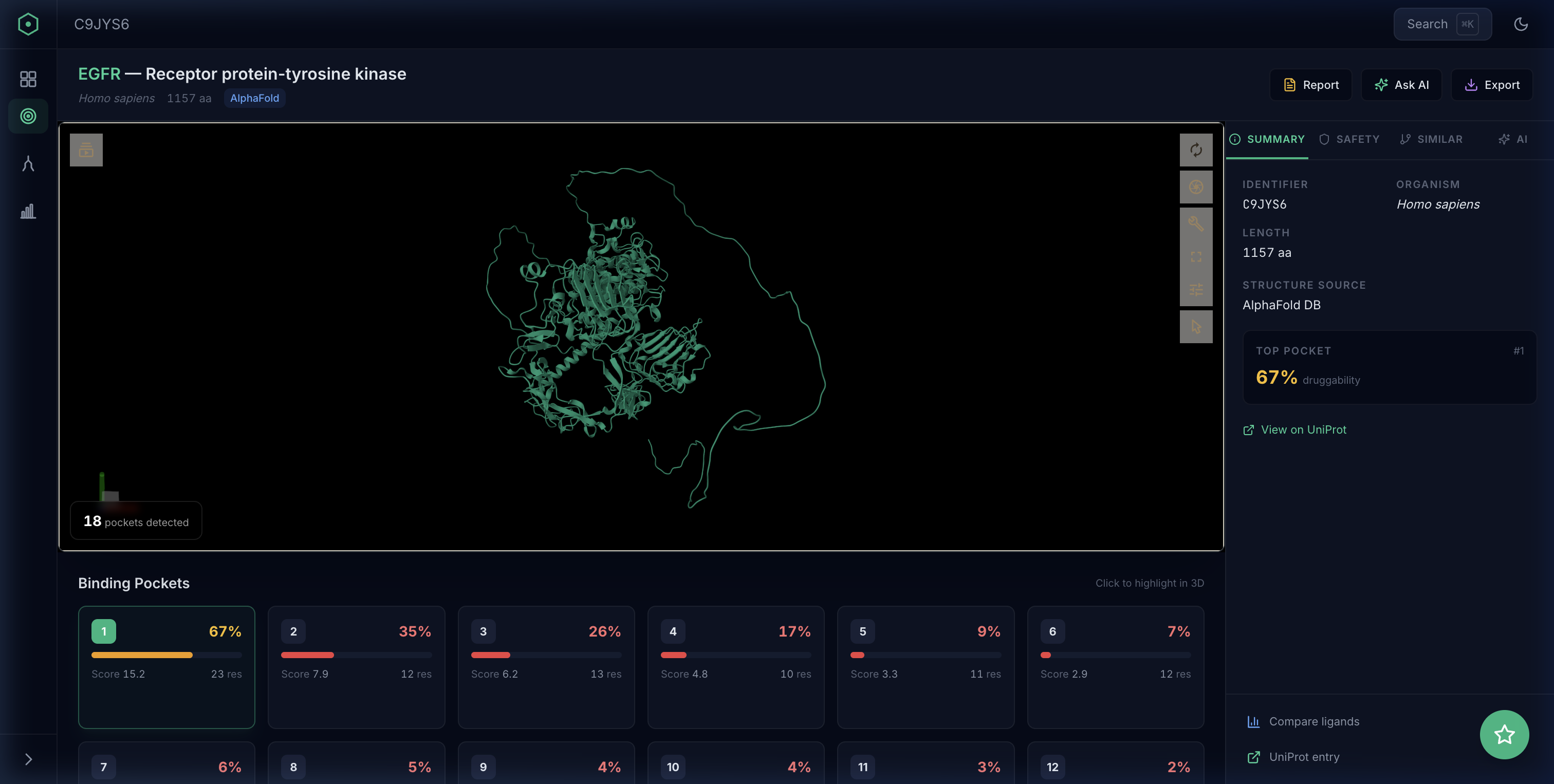
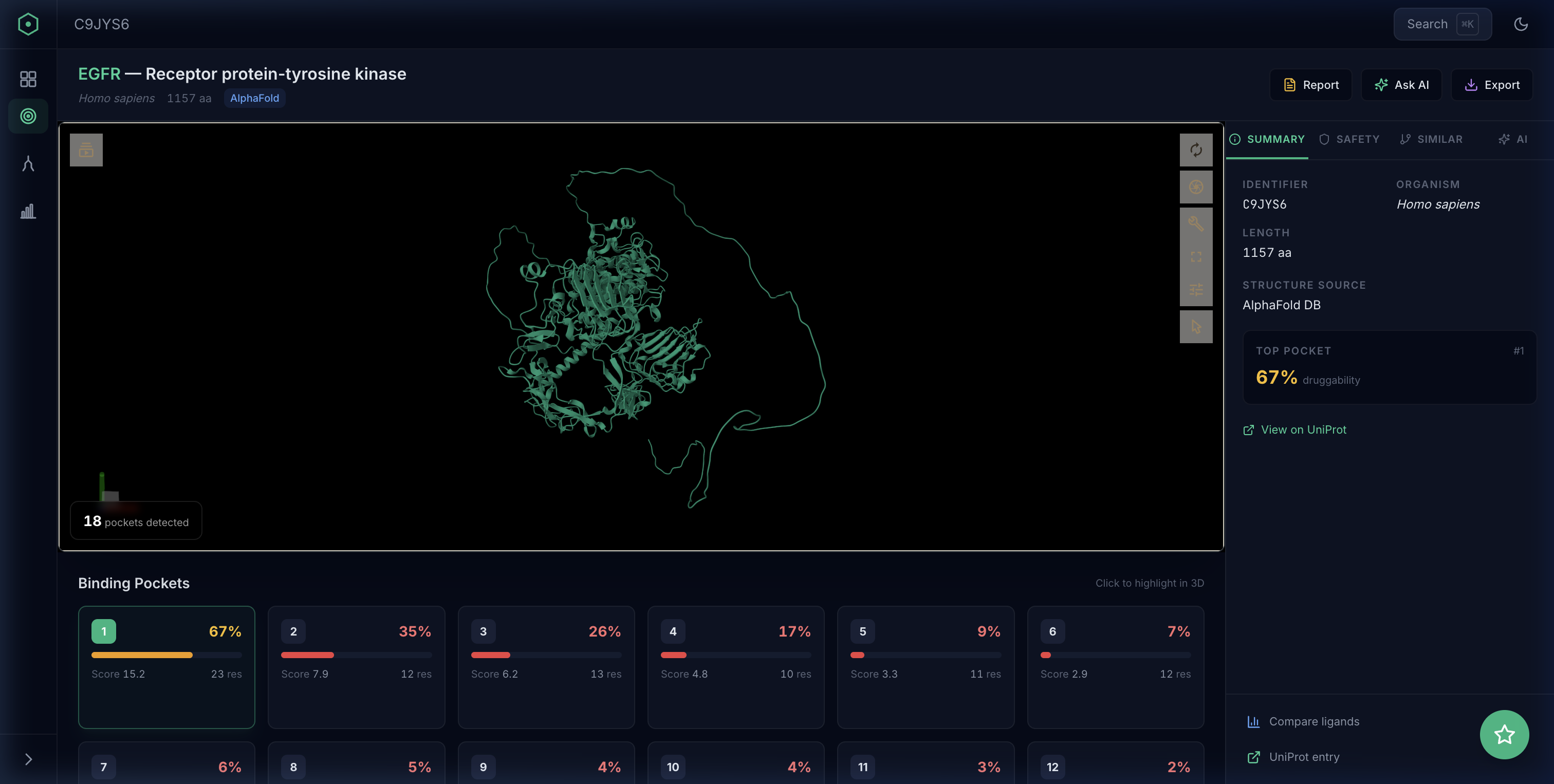
Pocket discovery
Enter any protein target and instantly see predicted binding pockets ranked by druggability. P2Rank’s ML algorithm identifies sites that traditional methods might miss — including allosteric and cryptic pockets.
Demo
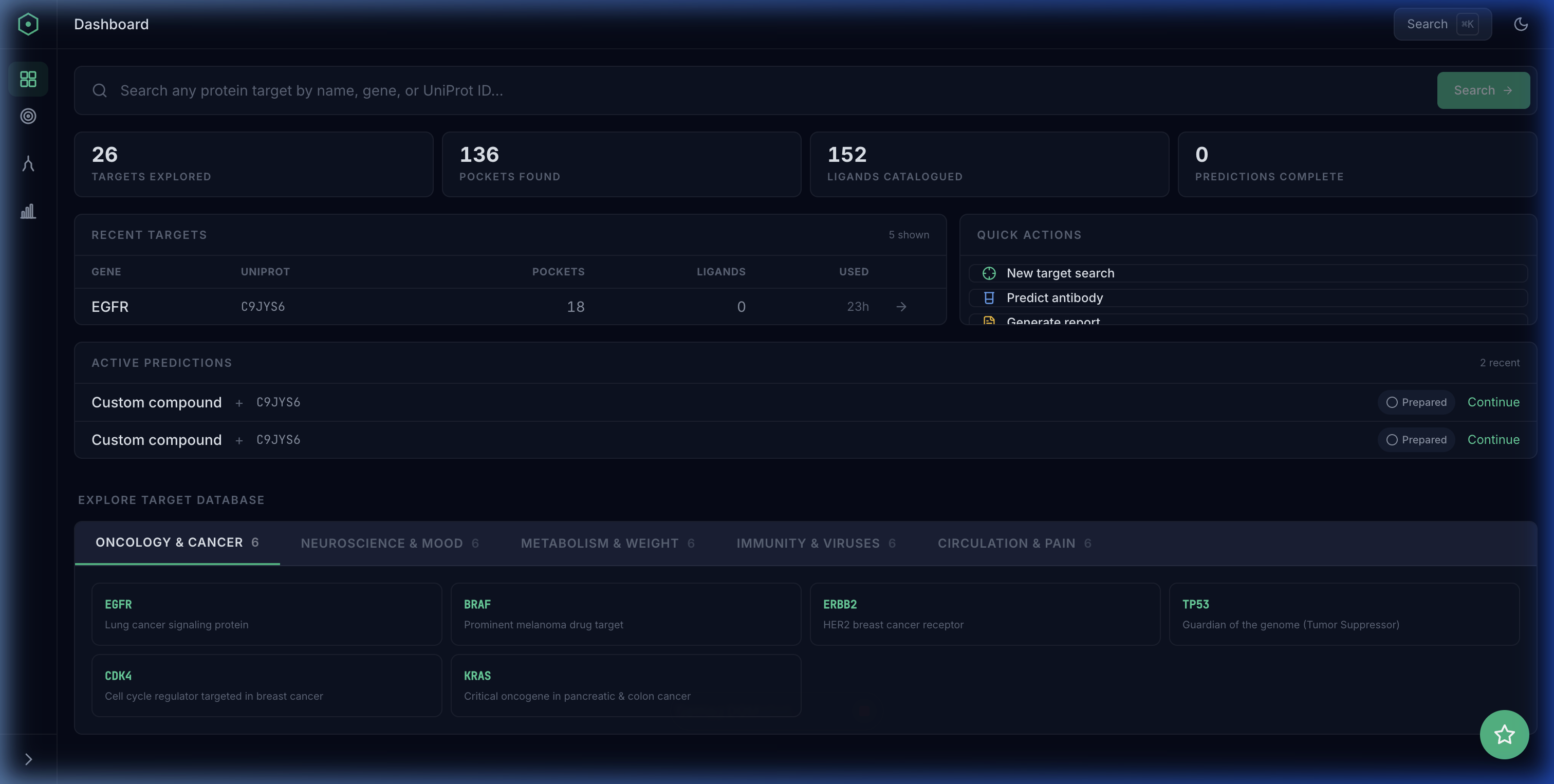
See it end to end
A guided walkthrough of the full OpenDDE pipeline — from target search to druggable insights — using real screenshots from the running app.
Use Cases
Who is OpenDDE for?
Academic researchers
Explore new drug targets for your research. Identify druggable pockets, find known compounds, and generate publication-ready druggability reports — all without commercial software licenses.
Explore targetsPharma scientists
Rapidly screen targets and validate druggability before committing lab resources. Compare pockets, analyze structure-activity relationships, and prioritize compounds computationally.
Start screeningStudents
Learn drug design concepts hands-on. Understand how binding pockets, ligand activity, and molecular predictions work in practice with real protein targets and compounds.
Start learningBiotech startups
Professional-grade target assessment without expensive commercial software. Generate investor-ready druggability reports with pocket analysis, ligand landscape, and AI-powered insights.
Assess targetsBuilt on giants
Powered by the best open-source tools
OpenDDE integrates world-class computational biology tools into a single, cohesive platform.
AlphaFold 3
Structure prediction
Google DeepMind
Boltz-2
Affinity prediction
MIT Jameel Clinic
P2Rank
Pocket detection
Czech Technical University
ChEMBL
Bioactivity data
EMBL-EBI
RDKit
Cheminformatics
Open-source community
ImmuneBuilder
Antibody modeling
Oxford Protein Informatics
UniProt
Protein knowledge base
UniProt Consortium
PubChem
Chemical data
NCBI / NIH
Claude AI
Scientific reasoning
Anthropic
Our mission
Open source and free forever
We believe computational drug design should be accessible to every researcher, not locked behind expensive licenses or proprietary platforms.
MIT Licensed
Use, modify, and distribute freely. No strings attached, no usage limits, no hidden fees.
Docker Ready
One command to run everything. Six containers, fully orchestrated, with health checks and auto-restart.
Community Driven
Built in the open. Every feature request, bug report, and pull request makes the platform better for everyone.
Standing on shoulders
Inspired by the frontier
OpenDDE draws deep inspiration from Isomorphic Labs' IsoDDE, which demonstrated that AI-first drug design can more than double the accuracy of existing methods in predicting binding poses. While IsoDDE remains a proprietary, frontier system, OpenDDE aims to bring similar workflows to the open-source community by composing the best freely available tools into a cohesive, Docker-native platform.
OpenDDE is an independent, community-built project and is not affiliated with Isomorphic Labs or Google DeepMind. Its modular architecture allows each component to be swapped as better open-source alternatives emerge.
Everything you need for drug design
From target identification to lead optimization, OpenDDE provides a complete computational drug design workflow.
Pocket Discovery
Machine-learning binding site prediction with P2Rank. Identifies druggable pockets with residue-level detail.
Ligand Intelligence
Fetch known drugs from ChEMBL with IC50, Ki activity data, clinical phase, and structure-activity relationships.
Complex Prediction
Generate AlphaFold 3 input for protein-ligand binding. Semi-automated prediction workflow.
Affinity Prediction
Boltz-2 predicts pIC50, IC50, and binder probability with iPTM/pLDDT confidence. Compare predicted vs experimental ChEMBL values side-by-side.
Virtual Screening
Run Boltz-2 across hundreds of ligands as a single campaign. Live progress, ranked top hits, CSV export, persistent results.
Antibody Modeling
Predict antibody 3D structures from VH/VL sequences using ABodyBuilder2 with CDR visualization.
AI Assistant
Claude-powered drug design insights. Context-aware analysis of targets, pockets, and ligands.
Analytics & Reports
Druggability reports, SAR plots, activity cliffs, pocket comparison, and PDF export.
Ready to explore?
Search any protein target and discover its druggable pockets, known compounds, and binding predictions in minutes.